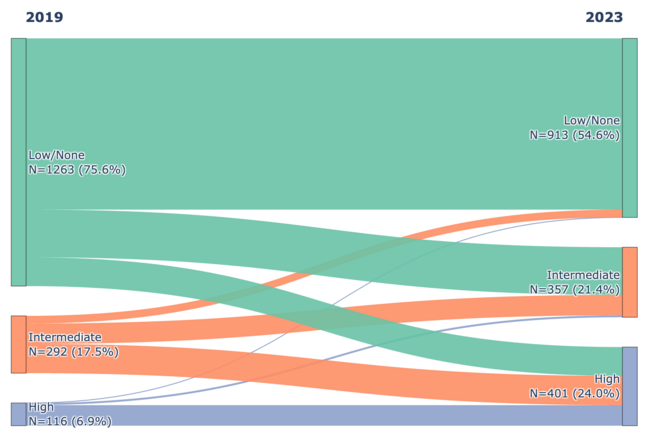
Recurrent Coronary Artery Thrombus Formation in the Setting of Diabetic Ketoacidosis
ABSTRACT: A 34-year-old male with insulin-dependent type II diabetes mellitus developed recurrent ST-elevation myocardial infarction in the setting of diabetic ketoacidosis. In both circumstances, coronary angiography revealed a large thrombus without angiographically evident coronary atherosclerosis. To our knowledge, this is the first report of diabetic ketoacidosis associated with coronary thrombosis in the absence of underlying atherosclerotic disease, inherited hypercoagulable disorders or previous personal or family history of thromboembolism.
Editor’s Note: “This complex case underscores the importance of metabolic abnormalities such as diabetic ketoacidosis contributing to recurrent coronary thrombosis. Hence, besides treating the coronary thrombus, strict control of metabolic abnormalities is also required. In addition, dual antiplatelet therapy of aspirin and clopidogrel is usually needed and may have prevented second presentation of coronary thrombosis." — Samin K. Sharma, MD, Mount Sinai Medical Center, New York, New York
Myocardial infarction in the setting of diabetic ketoacidosis (DKA) is well described and usually due to atherosclerotic disease. Rarely, it is associated with inherited hypercoagulable disorders.1 We present a case of recurrent acute coronary thrombus formation due to a hypercoagulable state in the setting of DKA but in the absence of angiographically evident atherosclerosis.
Case Report. A 34-year-old man with type II insulin-dependent diabetes mellitus, who was poorly compliant with medical therapy, presented to the emergency department with a 1-day history of nausea, vomiting and abdominal pain. He described the pain as diffuse, constant, and sharp and precipitated by vigorous vomiting with no relieving factors. On admission, he denied fever, chills, change in bowel movements, dysuria, hematuria or flank pain. He also denied any cardiac symptoms such as dyspnea, orthopnea, paroxysmal nocturnal dyspnea (PND), palpitations, presyncope or syncope. His clinical presentation was similar to that during previous presentations with diabetic ketoacidosis (DKA). Past medical history is as noted above. At home, he was prescribed humalog 75/25 30 units twice daily and regular insulin as per sliding scale, but had not taken these medications for 3 days. He had a 10-pack year history of smoking, without a history of illicit drug use. Family history was negative for premature coronary artery disease, stroke and thromboembolism.
On physical examination, his temperature was 97.7 °F, respiratory rate was 18 breaths/minute, pulse was 103 beats/minute, and blood pressure was 140/88 mmHg. His lungs were clear on auscultation and his cardiac exam revealed normal S1 and S2 without murmur, rubs or gallops. There were no carotid bruits. Jugular venous pressure was normal. Abdominal exam was notable for generalized tenderness without focality, guarding or rigidity. Lower extremity pulses were symmetrical and there was no edema.
Laboratory evaluation on presentation revealed a serum glucose of 653 mg/dl, an  anion gap of 36, a serum osmolality of 305 MOSM/L, arterial pH of 7.1, ketonemia and ketonuria. Complete blood count, liver function test, amylase, lipase, electrolytes and renal function test were within normal limits. His glycosylated hemoglobin was 14%. He was admitted with a diagnosis of DKA secondary to non-compliance with his insulin regimen. Within 6 hours of hospitalization, he developed severe stabbing chest pain at rest that was described as substernal, non-radiating and without associated symptoms. An electrocardiogram (ECG) demonstrated ST elevations in
anion gap of 36, a serum osmolality of 305 MOSM/L, arterial pH of 7.1, ketonemia and ketonuria. Complete blood count, liver function test, amylase, lipase, electrolytes and renal function test were within normal limits. His glycosylated hemoglobin was 14%. He was admitted with a diagnosis of DKA secondary to non-compliance with his insulin regimen. Within 6 hours of hospitalization, he developed severe stabbing chest pain at rest that was described as substernal, non-radiating and without associated symptoms. An electrocardiogram (ECG) demonstrated ST elevations in  leads II, III, aVF and V3–V6 (Figure 1). He was given 325 mg of aspirin and intravenous nitroglycerin, and underwent emergent coronary angiography. A small area of haziness suspicious for thrombus was noted in the ostial left anterior descending artery (LAD) followed by total occlusion distally (Figure 2). The remaining coronary arteries were normal in appearance. The ejection fraction (EF) was estimated to be 25% with antero-apical and diaphragmatic akinesis. Intravenous heparin and eptifibatide were administered. Large quantities of thrombus were removed with aspiration thrombectomy and Thrombolysis In Myocardial Infarction (TIMI) 3 flow was restored to the apex, but a mild filling defect persisted in the ostial LAD. An intravascular coronary ultrasound confirmed a small thrombus in the ostial LAD. Intravenous heparin and eptifibatide drips were continued for 48 hours and he was administered daily aspirin, carvedilol, lisinopril and simvastatin. His post-procedure CK and CK-MB were 1,966 IU/L and 177 ng/ml, respectively, which continued to trend down. A transesophageal echocardiogram did not reveal valvular vegetation or left ventricular (LV) or left atrial appendage thrombus. A follow-up coronary angiogram 2 days later demonstrated a small, persistent, non-flow limiting abnormality in the ostial LAD. The EF had improved to 40%. ECG at discharge demonstrated Q-waves in leads II, III and aVF, with poor R-wave progression. He was discharged on the above medications and counseled on the importance of adherence to his medical regimen. He was scheduled for an outpatient office visit with an endocrinologist.
leads II, III, aVF and V3–V6 (Figure 1). He was given 325 mg of aspirin and intravenous nitroglycerin, and underwent emergent coronary angiography. A small area of haziness suspicious for thrombus was noted in the ostial left anterior descending artery (LAD) followed by total occlusion distally (Figure 2). The remaining coronary arteries were normal in appearance. The ejection fraction (EF) was estimated to be 25% with antero-apical and diaphragmatic akinesis. Intravenous heparin and eptifibatide were administered. Large quantities of thrombus were removed with aspiration thrombectomy and Thrombolysis In Myocardial Infarction (TIMI) 3 flow was restored to the apex, but a mild filling defect persisted in the ostial LAD. An intravascular coronary ultrasound confirmed a small thrombus in the ostial LAD. Intravenous heparin and eptifibatide drips were continued for 48 hours and he was administered daily aspirin, carvedilol, lisinopril and simvastatin. His post-procedure CK and CK-MB were 1,966 IU/L and 177 ng/ml, respectively, which continued to trend down. A transesophageal echocardiogram did not reveal valvular vegetation or left ventricular (LV) or left atrial appendage thrombus. A follow-up coronary angiogram 2 days later demonstrated a small, persistent, non-flow limiting abnormality in the ostial LAD. The EF had improved to 40%. ECG at discharge demonstrated Q-waves in leads II, III and aVF, with poor R-wave progression. He was discharged on the above medications and counseled on the importance of adherence to his medical regimen. He was scheduled for an outpatient office visit with an endocrinologist.
Five months later, he presented to the ED with acute onset chest pain similar to his previous cardiac chest pain. The pain was associated with shortness of breath and started approximately 3 hours prior to presentation. There were no aggravating factors and it was relieved with 1 sublingual nitroglycerin administered in the emergency room. For the past week, the patient had been non-compliant with all of his medications. In addition, he was drinking 30 ounces of beer per day for the week prior to his admission.
Physical examination was unremarkable and no new ECG changes were noted.  Laboratory evaluation was consistent with DKA and the cardiac biomarkers were elevated (CK of 176 U/L, CK-MB of 14 ng/ml and troponin-I of 0.19 ng/ml). Within 5 hours of hospitalization, his chest pain recurred and was refractory to sublingual nitroglycerin. A repeat ECG revealed new ST-segment elevation in leads V3–V5 (Figure 3). An emergent coronary angiogram demonstrated a large filling defect in the proximal LAD at the takeoff of the first major diagonal branch, which narrowed the vessel lumen by 90% (Figure 4). The distal LAD was occluded without filling by collaterals. The
Laboratory evaluation was consistent with DKA and the cardiac biomarkers were elevated (CK of 176 U/L, CK-MB of 14 ng/ml and troponin-I of 0.19 ng/ml). Within 5 hours of hospitalization, his chest pain recurred and was refractory to sublingual nitroglycerin. A repeat ECG revealed new ST-segment elevation in leads V3–V5 (Figure 3). An emergent coronary angiogram demonstrated a large filling defect in the proximal LAD at the takeoff of the first major diagonal branch, which narrowed the vessel lumen by 90% (Figure 4). The distal LAD was occluded without filling by collaterals. The  remainder of the coronary vasculature was normal. A left ventriculogram revealed apical akinesis with an estimated EF of 40%. Aspiration thrombectomy yielded macroscopic white and red thrombus. A subsequent echocardiogram showed inferior and apical akinesis and an EF of 40% with a 1.1 x 0.8 cm LV apical thrombus.
remainder of the coronary vasculature was normal. A left ventriculogram revealed apical akinesis with an estimated EF of 40%. Aspiration thrombectomy yielded macroscopic white and red thrombus. A subsequent echocardiogram showed inferior and apical akinesis and an EF of 40% with a 1.1 x 0.8 cm LV apical thrombus.
On his second presentation, a hypercoagulable work-up had been completed prior to initiation of anticoagulation therapy (Table 1). This revealed a low normal protein C activity, low protein S activity and normal antithrombin III (AT III) antigen level. The von Willebrand  factor (vWF) activity, Factor VIII activity and fibrinogen level were elevated. Lupus anticoagulant, activated protein C resistance and anticardiolipin antibodies were negative. Homocysteine level was normal and prothrombin 2021A gene mutation was not present.
factor (vWF) activity, Factor VIII activity and fibrinogen level were elevated. Lupus anticoagulant, activated protein C resistance and anticardiolipin antibodies were negative. Homocysteine level was normal and prothrombin 2021A gene mutation was not present.
The patient was restarted on his previous medication regimen along with clopidogrel. He was also started on warfarin bridged with heparin given his LV apical thrombus.
Discussion. Diabetes mellitus (DM) creates a hypercoagulable state.2,3 In patients with DM, coagulation factors such as Factor VIII, fibrinogen and vWF are chronically elevated, predisposing them to a prothrombotic state.2,3 Elevation in these coagulation factors was also seen in our patient. In addition, a study comparing diabetic patients with non-diabetic patients reported acquired protein S deficiency in patients with diabetes mellitus.4 Our patient also had low levels of protein S activity. It has been shown in diabetics that they exhibit suppressed nitric acid and prostacyclin synthesis, leading to loss of their anti-aggregatory effects.3 These anti-aggregatory effects along with endothelial dysfunction increase platelet activation.5 All these homeostatic alterations lead to a persistent hypercoagulable state in diabetics.
An enhancement of this pre-existing prothrombotic state occurs during the initial 24 hours of onset of DKA when protein C activity and protein S activity further decrease. Decline in these levels likely causes an increase in the vWf Ag and FVIII levels, ultimately increasing the risk of vascular thrombosis.5 Our patient had a low normal protein C activity as well as elevated vWf Ag and FVIII levels, in addition to low protein S activity.
Fitzgerald first described arterial thrombosis in the setting of DKA in 1961.6 In his series of 160 cases with DKA, 19 deaths were reported, of which 5 were due to arterial thrombosis. The particular vessels reported in this case series included the aorta, iliac, subclavian, internal carotid and major branches of the renal and splenic arteries. Since then, arterial thrombosis of mesenteric, aortoiliac and femoral arteries has also been reported in the setting of DKA.7,8 Our case is the first reported case of recurrent coronary thrombus formation in the setting of DKA with no evidence of angiographically visible atherosclerotic disease. While it is likely that DKA led to a hypercoagulable state that facilitated thrombus formation, we cannot rule out the possibility that the MI was the cause rather than the result of DKA.
Conclusion. In our patient with recurrent ST-elevation MI who had no evidence of atherosclerotic disease or of an inherited hypercoagulable state, we suspect that exacerbation of pre-existing hypercoagulable tendencies seen in diabetics with DKA was responsible.
References
1. Doggen CJ, Cats VM, Bertina RM, Rosendaal FR. Interaction of coagulation defects and cardiovascular risk factors: Increased risk of myocardial infarction associated with factor V leiden or prothrombin 20210A. Circulation 1998;97:1037–1041.
2. Grant PJ. Diabetes mellitus as a prothrombotic condition. J Intern Med 2007;262:157–172.
3. Carr ME. Diabetes mellitus: A hypercoagulable state. J Diabetes Complications 2001;15:44–54.
4. Ceriello A, Giugliano D, Quantraro A, et al. Possible role for increased C4b-binding protein level in acquired protein S deficiency in type I diabetes. Diabetes 1990;39:447–449.
5. Carl GF, Hoffman WH, Passmore GG, et al. Diabetic ketoacidosis promotes a prothrombotic state. Endocr Res 2003;29:73–82.
6. Fitzgerald MG, O’Sullivan DJ, Malins JM. Fatal diabetic ketosis. Br Med J 1961;1:247–250.
7. Hamblin PS, Topliss DJ, Chosich N, et al. Deaths associated with diabetic ketoacidosis and hyperosmolar coma. 1973–1988. Med J Aust 1989;151:439–444.
8. Zipser S, Kirsch CM, Lien C, et al. Acute aortoiliac and femoral artery thrombosis complicating diabetic ketoacidosis. J Vasc Interv Radiol 2005;16:1737–1739.
From the *Department of Internal Medicine, Saint Joseph Mercy Hospital, Ann Arbor, Michigan and §Michigan Heart, Ypsilanti, Michigan. This abstract was presented at the Society of General Internal Medicine 33rd Annual Meeting, Minneapolis, Minnesota on April 29, 2010 and the American College of Physicians Associates Meeting (Michigan Chapter), Kalamazoo, Michigan on May 15, 2010. The authors report no conflicts of interest regarding the content herein. Manuscript submitted April 13, 2010, provisional acceptance given April 23, 2010, final version accepted May 25, 2010. Address for correspondence: Herbert D. Aronow, MD, MPH, Michigan Heart, P.C., 5325 Elliott Dr., Ste. #202, Ypsilanti, MI 48197. E-mail: haronow@michiganheart.com