News from the Transcatheter Cardiovascular Therapeutics Meeting (TCT)
November 2005
ACIST Medical Systems Introduces the ACIST CVi at the TCT
ACIST Medical Systems, a clinical and technological company in automated contrast delivery systems and part of the Bracco Group, announced the introduction of the ACIST CVi Contrast Delivery System for cardiovascular angiographic procedures. This product combines all of the features of the ACIST CMS and Voyager systems onto one platform. The ACIST CVi is expected to be commercially available in the first quarter of 2006, or before. This fourth generation product enhances the ease of contrast and saline injection for physicians, nurses and technicians with the following new system benefits:
ACIST CVi provides one contrast delivery system for both cardiac and vascular angiography. Synchronization capabilities are part of this integrated system.
The lower-profile, table-mountable injector and extendable control panel can be placed where it is needed most and with less interference. The ACIST CVi system can help reduce the amount of equipment in the procedure room and has features which shorten the learning curve for new users.1
ACIST CVi Fluid Delivery System continues to provide variable-rate adjustments of contrast flow and volume, in real-time, by adjusting the pressure applied to the ergonomic AngioTouch® hand controller. It offers cost effectiveness and enhanced workflow through the reduction of contrast media usage and needed set-up time.
Since the system's introduction in 1998, numerous articles in top-tier interventional cardiology journals have described its potential benefits. 1-6
1. J Invas Cardiol 2004; 16:360-362.
2. J Invas Cardiol 2005; 16:360-362.
3. Cath Lab Digest 4:15 suppl Oct 2002.
4. J Int Cardiol. 2001:12:147-152.
5. J Invas Cardiol 2005; 17:118-121.
6. J Invas Cardiol 2005, 16:729-731.
Taxus Express2 Stent System Shows Significant Reduction of Restenosis in Diabetic Patients in Clinical Trial
Boston Scientific Corporation announced nine-month sub-population data from its TAXUS V clinical trial that further supports the performance of the Taxus Express2 paclitaxel-eluting coronary stent system for the treatment of coronary artery disease in diabetic patients, particularly higher-risk, insulin-requiring diabetics. TAXUS V expands on the TAXUS IV pivotal trial by studying the most challenging lesions and highest-risk patients ever studied in a randomized, controlled clinical trial in the U.S.
"The TAXUS V diabetic sub-population data provides impressive results on of the outcome of the TAXUS stent system in treating complex patients, especially insulin-requiring diabetics," said Naim Z. Farhat, MD, who presented the results at TCT. "Most significantly, the data from this study shows an observed increasing benefit of the TAXUS system in the higher-risk insulin sub-group as compared to control and other diabetic patients."
The diabetic sub-population analysis demonstrated significant improvements among diabetic patients receiving the Taxus paclitaxel-eluting system versus those in the bare metal control group, and further improvements among the higher-risk, insulin-requiring diabetic patients. The nine-month target lesion revascularization rate (TLR) for the medically-treated diabetic sub-population of the TAXUS stent group was 9.6 percent compared with 17.5 percent in the control group (P=0.04), a reduction comparable to the non-diabetic sub-population (45 percent in both cases). Among insulin-requiring diabetic patients, the TLR rate was 8.3 percent compared to 17.3 percent in the control group (P=0.24), a reduction of 52 percent.
The diabetic sub-population also reported an in-segment binary restenosis rate of 18.2 percent in the TAXUS stent group compared with 38.4 percent in the control group (PClinical Trial Demonstrates Positive Results for St. Jude Medical Device Treating Coronary Bypass Graft Blockages
Data from a clinical trial show that a new embolic protection device from St. Jude Medical, Inc. demonstrated positive results for treating patients with saphenous vein coronary bypass grafts. The Proxis Embolic Protection System is currently under review by the U.S. FDA as an embolic protection system. Currently, Proxis is available in Europe for embolic protection during PCI for saphenous vein grafts. In the U.S., Proxis is cleared for Flow Control use, where a cardiologist can temporarily stop blood flow to a vessel and inject or aspirate fluid as needed.
The Proxis Embolic Protection System places a sealing balloon proximally to the lesion. The inflated balloon temporarily suspends blood flow so a guidewire can cross the lesion without sending debris into the bloodstream. An interventional device (such as a stent) is then delivered and the embolic debris is removed (aspirated) from the vessel. Once the intervention is complete, the balloon is deflated, blood flow is restored and Proxis is removed.
While several distal embolic protection devices are available, Proxis is the first-of-its-kind designed to treat a majority of the nearly 148,000 saphenous vein graft interventions in the U.S. each year (270,000 worldwide), including the nearly 20 percent of cases where current distal embolic protection devices cannot be used.
"Many vein graft lesions have challenged interventional cardiologists because their location has made it difficult, if not impossible, to position current embolic protection devices so as to protect the downstream bed," said Campbell Rogers, MD, Brigham and Women's Hospital, Boston, Mass., and Principal Investigator of the PROXIMAL Embolic Protection Trial who presented the study results at TCT. "The Proxis device not only allows protection for most vein graft lesions (and our results suggest it may offer better protection from post-procedural complications than current devices), but it also can be deployed proximal to lesions not suitable to protection with current distal protection devices."
The PROMIXAL trial assessed whether Proxis was substantially equivalent to previously-cleared devices as determined by the rate of major adverse cardiac events (MACE) at 30 days, the primary endpoint of the trial. Proxis proved equivalent to previously-cleared devices. In the as-treated population, meaning that patients were grouped depending on what type of embolic protection device they received, the MACE rate of the 241 patients treated with Proxis was 7.1 percent, compared to an 11.7 percent MACE rate in the 282 patients treated with the distal protection devices. In that same population, the study demonstrated that patients treated with Proxis were 60 percent less likely to experience a MACE than those treated with a distal embolic protection device. Moreover, when comparing device to device in the middle section of the vessel, where either a proximal or distal system could be used, the MACE rate for Proxis was 6.2 percent compared to 11.2 percent for the distal devices.
The trial began in June 2003 and involved 600 patients in a prospective, randomized, two-arm trial at 67 research sites nationwide. Patients scheduled to undergo a PCI on a saphenous vein graft lesion were randomized to a test arm or to a control arm. The MACE rate was defined as the composite of death, Q-wave or non-Q-wave myocardial infarction, emergent coronary artery bypass surgery (CABG), or target vessel revascularization (TVR).
Most Comprehensive U.S. Clinical Trial to Date Shows Competing DES Work Equally Well
After completing the most comprehensive U.S.-based study of its kind, Dr. Charles Simonton reported that the performance of two competing coronary stents was "virtually identical" in head-to-head comparisons. Simonton, a cardiologist associated with the Carolinas Heart Institute at Carolinas Medical Center, summarized his conclusions at the annual TCT (Transcatheter Cardiovascular Therapeutics) meeting in Washington, D.C.
The STENT registry, which now includes approximately 14, 000 patients, is the first of its kind to gather data from multiple sites, including eight medical centers located throughout the Southeast. The registry also includes one of the largest patient follow-up efforts based on a profile of the general populace. Earlier clinical trials focused on patient populations carefully screened to exclude high risk cases.
The new study, which involves non-proprietary research, shows outcomes the same for TVR (target vessel re-vascularization) and MACE (major adverse cardiac events, such as myocardial infarction, stroke or death, following interventional treatment).
"The present study shows that Taxus stents are being implanted in patients who are slightly older, have higher lesion risk and somewhat smaller vessels than patients receiving Cypher stents," said Dr. Simonton. "Based on data collected so far, paclitaxel- and sirolimus-eluting stents demonstrate similar efficacy and safety overall at nine months. When you adjust the two groups to account for the higher complexity level of the Taxus stent arm, results favor the Taxus stent, but without statistically significant differences between the two stents."
Eight coronary interventional centers across the United States are participating in the registry, which will enroll more than 8,000 patients, the largest population in any study of its kind. A total of 3,758 patients receiving either the Taxus stent (1,476) or the Cypher stent (2,282) had completed nine months of follow up at the time of the interim analysis. The majority of baseline characteristics were similar among the two treatment groups, although Taxus stent patients tended to be slightly older with more acute coronary syndrome, slightly smaller vessel diameters and higher ACC lesion risk scores. Late clinical outcomes trend numerically in favor of paclitaxel as compared to sirolimus, including death (2.1 vs. 2.7), myocardial infarction (1.8 vs. 2.2), TVR (3.4 vs. 4.2), and MACE (6.8 vs. 7.9). In addition, both the Taxus and Cypher stents showed low rates of thrombosis that are consistent with results seen in bare-metal stents.
The STENT registry is a partnership of the Dickson Institute for Health Studies at Carolinas HealthCare System (CHS), the Sanger Clinic (a CHS-owned practice where Dr. Simonton is employed), and the Carolinas Heart Institute (a specialized coronary care unit at Carolinas Medical Center).
Most of the early FDA-sanctioned studies relied on data available three or six months post-procedure. The STENT registry coordinated by CHS uses nine-month follow-up, and has a participation rate exceeding 95% of eligible patients. Additional data from STENT are scheduled to be presented at the American College of Cardiology conference in 2006, with the final analysis slated for presentation at TCT 2006.
Three-year SAPPHIRE and U.S. Carotid Feasibility Trials Demonstrate Durability of Carotid Stenting
Preliminary three-year data from the SAPPHIRE and final three-year data from the U.S. Carotid Feasibility Study (USFS), presented at the 2005 TCT meeting, demonstrate the long-term durability of carotid artery stenting (CAS) for the prevention of stroke versus carotid endarterectomy (CEA) in high risk surgical patients.
Three hundred thirty-four patients in the SAPPHIRE trial were randomized to either CAS or CEA. Patients who received CAS with the Precise® Nitinol Self-Expanding Stent and Angioguard XP Emboli Capture Guidewire System maintained a low incidence of stroke after the first 30 days for the duration of the three-year follow-up in the USFS and SAPPHIRE. In the SAPPHIRE trial, at 36 months the incidence of stroke was virtually identical for both CAS and CEA (7.1% for CAS and 6.7% for CEA, P = 0.945). The 3-year incidence of stroke across the randomized and non-randomized CAS treatment groups in SAPPHIRE and the USFS had only an average increase of 4.0% over the 30-day stroke rate. Cordis Corporation, a Johnson & Johnson company, sponsored the SAPPHIRE and USFS clinical trials.
Jay S. Yadav, MD, of the Cleveland Clinic Foundation, and a principal investigator for the SAPPHIRE trial said, "The SAPPHIRE trial demonstrated CAS was non-inferior to CEA. This is important new data which suggests the long-term durability of CAS in this patient population."
An analysis of target lesion revascularization (TLR) with follow up to three years in the randomized arms of SAPPHIRE showed reintervention rates of 3.0% for CAS and 7.1% for CEA, P = 0.084. Three-year TLR rates for CAS were similar in the USFS.
Data from the SAPPHIRE, USFS, and CASCADE clinical trials also were presented in two poster sessions highlighting CAS involving Angioguard. The data suggests emboli protection is effective in preventing major strokes during carotid stenting. 30-day stroke rates were 8.6% for the 266 patients treated with stenting alone and 2.6% for the 116 patients treated by stenting with Angioguard. In addition, no patients treated with an Angioguard in CASCADE, USFS or in the treated randomized portion of SAPPHIRE had a major stroke. Approximately two-thirds of the minor ipsilateral strokes that occurred in these trials resolved with time.
Guidant Announces Positive Preliminary Results of Large Real-World Carotid Artery Stenting Study
Study Suggests Therapy Can Be Performed Safely by Broad Group of Physicians
Guidant Corporation announced preliminary results of its study of carotid artery stenting in high-risk patients, called CAPTURE (Carotid Acculink/Accunet Post Approval Trial to Uncover Rare Events). Results were presented at the TCT meeting in Washington, D.C. by Jay Yadav, MD from the Cleveland Clinic Foundation.
CAPTURE is an FDA-required post-approval study utilizing Guidant's carotid stent and embolic protection system. Key objectives of CAPTURE were to determine whether carotid artery stenting can be performed safely in real-world clinical settings with physicians of varying levels of experience and to evaluate the effectiveness of Guidant's training program.
The initial results of the study confirm positive earlier data on carotid stenting for high-risk patients. In addition, the study suggests that carotid artery stenting, which was introduced in the United States last year with the launch of Guidant's system, can be performed safely by physicians of different experience levels. The rate of death, stroke and myocardial infarction within 30 days of the procedure was 5.1 percent.
"CAPTURE is a landmark study. It is the largest and most rigorous real-world study of carotid stenting with independent neurologic follow-up and clinical events adjudication. It demonstrates, for the first time, that with proper training carotid stenting can be performed with exemplary results by a variety of physicians in community hospital settings," said Dr. Yadav. "The excellent results and the overall ease of use of the system make this a breakthrough treatment for stroke prevention in high-risk patients."
Results were based on data from 1,603 patients receiving Guidant's FDA-approved RX Acculink Carotid Stent System and RX Accunet Embolic Protection System, which are indicated for high surgical risk patients. Patients in CAPTURE were treated by 240 physicians, including interventional cardiologists, interventional radiologists, interventional neuroradiologists, vascular surgeons and neurosurgeons, at 118 hospitals in the United States. CAPTURE has enrolled more than 2,800 patients to date and continues to enroll.
ARCHeR, the pivotal trial for the FDA approval of Guidant's carotid stent and embolic protection system, enrolled 581 patients at 48 centers with previous experience in carotid artery stenting. Results of the study demonstrated an 8.3 percent rate of death, stroke and myocardial infarction within 30 days of the procedure. ARCHeR enrollment was limited to high-risk patients. In addition to the CAPTURE and ARCHeR series of trials, Guidant is participating in the CREST trial evaluating carotid artery stenting in low-surgical risk patients, which continues to enroll.
Guidant's devices are available only to physicians with the appropriate training and prerequisite experience for performing carotid interventions. The company has worked closely with the FDA, medical societies and leading practitioners to develop targeted training programs in the use of the Acculink/Accunet Systems for physicians based on their experience level.
Cost Analysis Suggests Cypher Coronary Stent Cost Effective
An independent analysis of a clinical trial comparing the cost-effectiveness of the Cypher® Sirolimus-eluting Coronary Stent vs. bypass surgery suggests that treatment with the Cypher Stent offers a potential cost-savings over bypass surgery. The findings from the Arterial Revascularization Therapy Study (ARTS) I and II were presented at the annual TCT 2005 conference.
Treatment of multi-vessel coronary artery disease with the Cypher Stent was found to be substantially more cost-effective than bypass surgery both during initial hospitalization and at the one-year follow-up.
According to David J. Cohen, MD, MSc, Associate Director of Interventional Cardiology at Beth Israel Deaconess Medical Center in Boston, and Director of Cardiovascular Outcomes Research, Harvard Clinical Research Institute, who presented the results, "These findings provide additional evidence on the benefit of drug-eluting stents in treating complex patient cases that would have been directed to surgery," Dr. Cohen said. "Furthermore, the analysis shows that the economic benefit of the Cypher Stent technology is as significant as the clinical benefit we see in these challenging patient cases."
To account for baseline differences, both costs and event rates from ARTS-II were adjusted to reflect the number of a patient's diseased vessels and whether or not the patient had diabetes. Savings related to the avoidance of death, heart attack, revascularization and stroke were examined.
Initial hospitalization costs, accounting for physician fees, room and ancillary charges, and repeat procedures, were $7,700 less (29 percent) for Cypher Stent than for CABG, at $26,419 and $34,119 respectively.
Total one-year costs, when adjusted for patient differences in extent of coronary artery disease and diabetes, were $6,487 less (21 percent) for Cypher Stent than for CABG, at $30,388 and $36,875 respectively.
ARTS II is a single arm, randomized multi-center trial of 607 patients in 19 countries. The study sample was drawn from the ARTS-I randomized trial (1997-1998) and the ARTS-II registry (2003). Study criteria included patients with multivessel disease who required treatment of at least two major coronary arteries and who had stenoses that would be receptive to stenting.
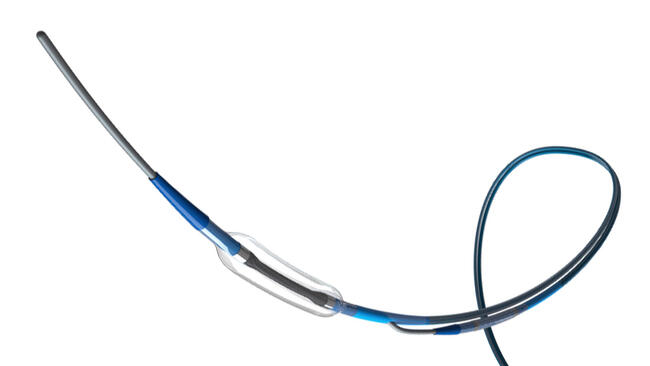
Evalve Announces Intiation of Trial of Percutaneous Alternative to Open-Heart Mitral Valve Surgery
Initial Clinical Study Results Are Also Presented at TCT
Evalve, Inc. announced the successful initiation of the study Endovascular Valve Edge-to-Edge REpair STudy (EVEREST) II, a randomized, multi-center study designed to demonstrate the safety and efficacy of its MitraClip percutaneous valve repair system for patients with mitral regurgitation (MR). EVEREST II will compare the MitraClip technique to standard surgical mitral valve repair or replacement in patients with functional or degenerative MR. Approximately 30 medical centers across North America will be participating in the study.
To qualify for the EVEREST II trial, patients must have moderate to severe or severe mitral regurgitation and be experiencing symptoms; or, lacking these symptoms, they must have a weakened left ventricle. The Phase II study is being conducted under a United States Food and Drug Administration (FDA) approved Investigational Device Exemption.
Positive long-term follow-up results from the company's initial clinical study, EVEREST I, were also presented at the TCT. The EVEREST I feasibility study has enrolled 47 patients with moderately severe or severe mitral regurgitation who were experiencing symptoms or had a weakened left ventricle. Only four percent of the 47 patients enrolled experienced a significant adverse event at 30 days. The first 27 patients treated have reached one-year follow-up. Ninety-three percent of those patients who experienced a significant reduction in MR at one month following treatment have maintained that improvement at one year. While all patients required surgery prior to the MitraClip procedure, 75 percent of those who received a clip remain free from surgery.
The MitraClip demonstrated it could reduce MR severity from moderate-severe or severe to moderate or lower. Importantly, the MR reduction achieved at 30 days is maintained at one year in 93% of patients and 74% of all patients treated have avoided open heart surgery.
This is a very exciting study because the only option currently available for patients with moderate to severe MR is open-heart surgery, said EVEREST principal investigator Ted Feldman, MD, director of the cardiac catheterization lab at Evanston Hospital in Evanston, IL. These initial results are very encouraging in that not only did we observe a reduction in MR at six months, but the durability of the repair has held up in those patients who have completed 12-month follow up.
Early-Phase Data Show Prasugrel Demonstrates More Consistent Platelet Inhibition Compared to Clopidogrel
Eli Lilly and Company and Sankyo Co., Ltd. announced data results at the TCT from three early-phase, cross-over studies demonstrating that all subjects responded to prasugrel, an investigational antiplatelet agent, while 22 to 43 percent of the same subjects did not respond to clopidogrel (Plavix), as measured by objectively defined parameters for inhibition of platelet aggregation. Prasugrel is an investigational antiplatelet agent designed to prevent platelet activation and aggregation by blocking adenosine diphosphate receptors on the platelet surface.
Eli Lilly and Sankyo Co., Ltd. are developing prasugrel as a potential treatment for patients who have suffered a heart attack or unstable angina. Recent studies suggest that a relationship may exist between a poor platelet response to antiplatelet agents in individual patients and poor clinical outcomes, which manifest as major adverse cardiovascular events, including heart attacks.
The pooled analysis, Clopidogrel Nonresponders: A Comparison With Prasugrel (CS-747, LY640315), a Novel Thienopyridine P2Y12 Receptor Antagonist, is based on the results of three, single-center, two-way cross-over clinical pharmacology studies. One hundred and twelve healthy volunteers aged 18-65 years old were randomized to receive either a 60 mg loading dose of prasugrel or the approved 300 mg loading dose of clopidogrel in a 2-way cross-over design. Adenosine diphosphate (ADP) was used to induce platelet aggregation in samples of subjects' blood. Inhibition of platelet aggregation (IPA) and the change in maximum platelet aggregation (MPA) from baseline were evaluated at both 4-5 and 24 hours after the medication was administered.
Nonresponders were objectively defined as subjects achieving less than 25 percent IPA or a difference of less than 20 percent in MPA in response to 5 µM adenosine diphosphate (ADP), and less than 20 percent IPA or a difference of less than 15 percent in MPA in response to 20 µM ADP. These thresholds represent levels of platelet inhibition that are more consistent with a placebo than that of an active antiplatelet agent.
All subjects responded effectively to a loading dose of prasugrel 60 mg. However, when the same subjects were administered 300 mg of clopidogrel, 22 percent were nonresponders based on IPA in response to 5 µM ADP (p<.001 and="" percent="" were="" nonresponders="" based="" on="" ipa="" in="" response="" to="" adp="" a="" phase="" iii="" clinical="" study="" with="" prasugrel="" triton-timi="" is="" underway.="" this="" head-to-head="" will="" compare="" the="" effects="" of="" clopidogrel="" up="" patients="" acute="" coronary="" syndrome="" who="" suffer="" heart="" attack="" or="" have="" unstable="" angina="" are="" undergo="" pci.="" primary="" focus="" two="" agents="" ability="" prevent="" stroke="" death="" secondary="" look="" at="" impact="" bleeding="" hospitalization="" for="" recurrent="" ischemia="" need="" urgent="" target="" revascularization.="" it="" anticipated="" that="" triton="" timi-38="" should="" be="" completed="" early="" regulatory="" submissions="" follow="" second="" half="">CoreValve's ReValving System Used Instead of Open-Heart Surgery to Non-Surgically Implant a New Aortic Bioprosthesis Over the Diseased Heart Valve
CoreValve announced that its second-generation, proprietary aortic heart valve was implanted successfully in a high-risk patient using CoreValve's percutaneous ReValving System during the opening session at the TCT. Eberhard Grube, MD, chief of cardiology at The Heart Center, Siegburg, Germany, and a consulting professor of medicine at Stanford University, and Jean-Claude Laborde, MD, an interventional cardiologist at Clinique Pasteur in Toulouse, France, performed the ReValving procedure in the cath lab at The Heart Center. As a result, their patient was able to avoid open-heart surgery to treat a defective aortic heart valve.
CoreValve's proprietary delivery system for percutaneous heart valve replacement is based on a catheter-and-self-expanding-frame approach on a beating heart, thus avoiding open-heart surgery. The CoreValve procedure - with the proprietary CoreValve Percutaneous ReValving System - can be performed in a cardiac cath lab just like angioplasty and stenting, resulting in less trauma to the patient and substantial cost-savings to the healthcare system.
The live ReValving procedure at TCT used CoreValve's own proprietary porcine pericardium aortic heart valve, delivered via CoreValve's small 21-French-sized catheter, and required less than 15 minutes to be implanted.
Taxus Clinical Trial Data Demonstrates Excellent Long-Term Safety & Efficacy Record
Boston Scientific Corporation announced follow-up data supporting excellent long-term safety and efficacy from four of its TAXUS clinical trials. The Company reported results from TAXUS I (four years), TAXUS II (three years), TAXUS IV (three years), and TAXUS VI (two years).
"The TAXUS clinical results reported today are outstanding and demonstrate the long-term safety and efficacy of paclitaxel-eluting stent technology," said Gregg W. Stone, MD, Principal Investigator of the TAXUS IV clinical trial and Professor of Medicine, Columbia University Medical Center in New York. "The TAXUS stent demonstrates a sustained and robust benefit in all subgroups examined out to three years in TAXUS IV, with no late catch-up apparent."
The TAXUS stent system has now established a long-term safety record as demonstrated by clinical trial data now reporting out to four years. TAXUS I, the longest running TAXUS clinical trial to date, reported safety results out to four years with no cases of stent thrombosis, death or myocardial infarction since patient enrollment. TAXUS II, the first TAXUS clinical trial to evaluate late clinical safety and efficacy outcomes of both the slow- and moderate-release formulations, reported no stent thrombosis from two to three years and a low overall cardiac death rate of 1.6 percent to three years. TAXUS IV, the United States pivotal trial, reported no new cases of stent thrombosis out to three years and a 1.1 percent cardiac death rate and 2.4 percent myocardial infarction rate from nine months to three years. TAXUS VI reported one case of stent thrombosis between one and two years with an overall 0.5 percent cardiac death rate.
The TAXUS clinical trials have reported long-term efficacy at three- to four-year follow-up as demonstrated by low target lesion revascularization (TLR) rates reported from TAXUS I, TAXUS II, TAXUS IV and TAXUS VI trials. There have been no cases of TAXUS TLR for the TAXUS I clinical trial out to four years. The long-term TAXUS II trial results suggest that the TAXUS stent system stably inhibits restenosis as demonstrated by significant reductions in TLR. TAXUS II trial reported no new cases of TLR in the TAXUS stent groups from two to three years with a low overall TLR rate of 5.4 percent in the slow-release formulation and 3.7 percent in the moderate-release formulation, as compared to 15.7 percent in the control (bare metal stent) group (p=0.0001), resulting in an absolute reduction of 10.3 percent versus control for the slow-release formulation and a 12 percent absolute reduction versus control for the moderate-release formulation. TAXUS IV trial featured markedly reduced restenosis resulting in lower rates of bypass graft surgery and repeat percutaneous interventions. The three-year TLR rate for TAXUS IV trial was 6.9 percent for the TAXUS stent group, as compared to 18.6 percent for the control group (P less than 0.0001), resulting in an absolute reduction of 11.7 percent versus control. In TAXUS VI, which used the moderate-release formulation, patients in the TAXUS stent group experienced a 54 percent reduction in TLR out to two years. The overall TLR rate in the TAXUS stent group of TAXUS VI was 9.7 percent, as compared to 21 percent for the control group out to two years (P(equal sign)0.0013). TAXUS IV and TAXUS VI are studying a more complex patient population than TAXUS I and TAXUS II.
The TAXUS I trial is a randomized, double-blind, multi-center, feasibility study designed to assess safety of the Company's slow-release drug- eluting stent platform. The trial was conducted at three centers in Germany and enrolled 61 patients. The TAXUS II trial is a 536-patient, 15-country, randomized, double-blind, controlled study of the safety and efficacy of a TAXUS paclitaxel-eluting coronary stent, in which two sequential cohorts of patients with standard risk, de novo coronary artery lesions were treated with different dose formulations. TAXUS IV is a randomized, double-blind pivotal trial designed to assess the safety and efficacy of a paclitaxel-eluting coronary stent system in reducing restenosis in de novo lesions 10 - 28 mm in length and 2.5 - 3.75 mm in diameter. The study enrolled 1,326 patients at 73 sites in the United States. The TAXUS VI trial is studying 448 patients with complex coronary artery disease at 44 sites. It is designed to establish the safety and efficacy of the moderate-release formulation TAXUS stent in the treatment of longer lesions. The TAXUS Express2 moderate-release paclitaxel- eluting stent is not approved for commercial distribution.
Toshiba Announces Launch of Five-Axis Infinix CF-I Cardiac Imaging System
Toshiba America Medical Systems, Inc. introduced the five-axis Infinix CF-i/SP (single plane) diagnostic and interventional imaging system, delivering head-to-toe and fingertip-to-fingertip patient access. Launched at TCT, the system was designed using Toshiba's Voice of the Customer feedback program, which utilizes clinician feedback to guide future product development.
Ronald P. Karlsberg MD, Clinical Professor of Medicine, David Geffen School of Medicine, UCLA, and Director of the Cardiac Catheterization Laboratory, Brotman Medical Center, commented, The development of five-axis technology sets a new standard for floor-mounted systems. With this system, even lower leg and radial access to patients is achieved quickly and easily for patients of all sizes. There is complete open access to the patient and even with the C-arm rotated to the side, the head is always up for proper anatomic orientation.
The floor-mounted Infinix CF-i/SP's five-axis design permits shifting of the C-arm around the exam table for angling. In addition, the Infinix CFi/SP is equipped with Toshiba's 8-inch cardiac flat panel detector (FPD) mounted on a fifth axis which rotates exactly as the C-arm rotates to the side. This fifth axis results in images always maintained in a heads-up orientation while the C-arm rotates.
Other system features include an anode heat-capacity liquid metal bearing tube of 3.0 MHU, which virtually eliminates overheating. The liquid metal tube allows for immediate image capture.
Configurations include single plane floor-mounted, single-plane ceiling-mounted, bi-plane and dual-plane flat panel detector systems.
NULL
Subscriber
Cath Lab Digest Newsletter