



The Trial Design of the Concurrent Optical and Magnetic Stimulation (COMS) Therapy Study for Refractory Diabetic Foot Ulcers (MAVERICKS):
A Multicenter, Randomized, Sham-Controlled, Double-Blind Investigational Device Exemption Clinical Study
A Multicenter, Randomized, Sham-Controlled, Double-Blind Investigational Device Exemption Clinical Study
© 2025 HMP Global. All Rights Reserved.
Any views and opinions expressed are those of the author(s) and/or participants and do not necessarily reflect the views, policy, or position of Wounds or HMP Global, their employees, and affiliates.
Abstract
Background. Diabetic foot ulcers (DFUs) are a major clinical challenge, particularly among patients with refractory ulcers, that often lead to severe complications such as infection, amputation, and high mortality. Innovations supported by strong clinical evidence have the potential to improve healing outcomes, enhance quality of life, and reduce the economic burden on individuals and health care systems. Objective. To describe the design of the concurrent optical and magnetic stimulation (COMS) therapy Investigational Device Exemption (IDE) study for refractory DFUs (MAVERICKS) trial. Materials and Methods. The MAVERICKS trial is a pivotal, multicenter, randomized, sham-controlled, double-blind study designed to evaluate the efficacy and safety of COMS therapy as an adjunct to standard of care for hard-to-heal DFUs. What differentiates MAVERICKS is its extended screening phase, ensuring the inclusion of truly refractory ulcers, thus addressing a critical gap in previous DFU research. With an expected enrollment of 224 patients across diverse clinical settings, the trial incorporates robust methodologies to eliminate bias, and comprehensive inclusion and exclusion criteria to ensure data integrity. The study’s primary outcome measure, time to complete wound closure, as well as secondary end points including wound area reduction and time to amputation, will provide credible insights into the therapeutic potential of COMS. Conclusion. The MAVERICKS trial is particularly significant for investigating a novel, accessible, and cost-effective approach to advance DFU treatment. This trial sets a high standard in DFU research and seeks to provide reliable evidence to improve patient outcomes and guide future clinical practice. Upon trial completion, the authors look forward to sharing the findings and outcome analysis with wound care stakeholders. The study is registered under ClinicalTrials.gov identifier NCT05758545, effective March 7, 2023.
The successful treatment of Wagner grade 1 and 2 diabetic foot ulcers (DFUs) represents the most common pathway to US Food and Drug Administration (FDA) clearance for use of a device in the chronic wound space. This population of patients is abundant and continues to have suboptimal outcomes. In addition, the general model for prospective randomized trials in this population of patients is well defined.
The investigators are therefore undertaking an FDA Category B investigational device exemption (IDE) clinical trial to evaluate the efficacy and safety of concurrent optical and magnetic stimulation (COMS) therapy in the treatment of refractory DFUs. Given that wound healing studies of this magnitude and design quality are valued, the authors have chosen to highlight and support the protocol construction and plan for this clinical trial, which is currently enrolling patients. The authors look forward to sharing the findings and outcome analysis of this trial with wound care stakeholders following study completion.
COMS therapy
The chronicity of DFUs can be attributed to multiple impaired physiologic mechanisms at the cellular, molecular, and tissue levels. Excess inflammatory stimuli or immune cells can prevent the wound from progressing to the proliferative phase of healing, while impaired tissue perfusion and abnormal angiogenesis result in oxygen deprivation and reduced cellular adenosine triphosphate (ATP) synthesis.1-3 Prior research has demonstrated that enhancing cellular energy can significantly accelerate healing through improved platelet accumulation, and upregulation of cytokine production, stem cells, and vascular endothelial growth factor.3,4
Physical energy-based devices, such as electrical stimulation, electromagnetic therapies, ultrasound, and extracorporeal shockwave therapy, have shown promise in restoring wound healing capacity but lack widespread adoption due to issues including portability and reliance on specialist clinicians.5,6
A novel technology combines concurrent optical and magnetic stimulation to deliver synergistic and supra-additive effects at the cellular level. Magnetic stimulation modulates intracellular calcium, enhancing nitric oxide production, tissue perfusion, and angiogenesis, while optical stimulation boosts mitochondrial respiration and ATP production.7-9 This dual-modality approach, targeting both perfusion and cellular energetics, represents a significant advancement in wound care technology based on physical energy delivery. By addressing the dynamic and synergistic aspects of wound healing at the molecular and cellular levels, this technology offers a potential breakthrough for treating chronic DFUs.
The COMS One therapy system (hereafter “COMS system”), developed and manufactured by Piomic Medical AG, addresses these limitations with a portable and minimal maintenance design, enabling use across different care settings, from outpatient wound clinics to home health. This care setting flexibility, rarely associated with advanced wound care devices, is particularly beneficial to patients experiencing mobility or transportation difficulties, which are common obstacles to regular appointment attendance and consistent care, and thus primary contributors to poor healing outcomes. The COVID-19 pandemic exacerbated these issues, forcing wound care specialists to revise treatment regimens and involve less-skilled caregivers, such as family members, in providing general wound care. This highlighted the need for advanced wound care modalities that can be reliably delivered across various care settings by a wide range of caregivers.
The COMS system administers a 16-minute treatment using the COMS One device (hereafter “capital equipment” or “capital device”) and 2 single-use disposable components. The COMStouch (Piomic Medical AG) disposable coupling device is an interface between the capital device and the patient’s skin. The coupling device is manufactured using sterile silicone for prevention of cross-contamination and infection, and for its softness and flexibility for patient comfort and positioning ease. The coupling device attaches to the capital device to form a single, integrated treatment assembly. The non-sterile COMSfix (Piomic Medical AG) disposable component is a hook-and-loop fixation device that secures the integrated treatment assembly to the patient during treatment. Rechargeable and portable, the COMS system integrates into all care settings while preserving established standard of care (SOC) regimens. Figure 1 depicts the COMS system, including the capital equipment and related consumables.
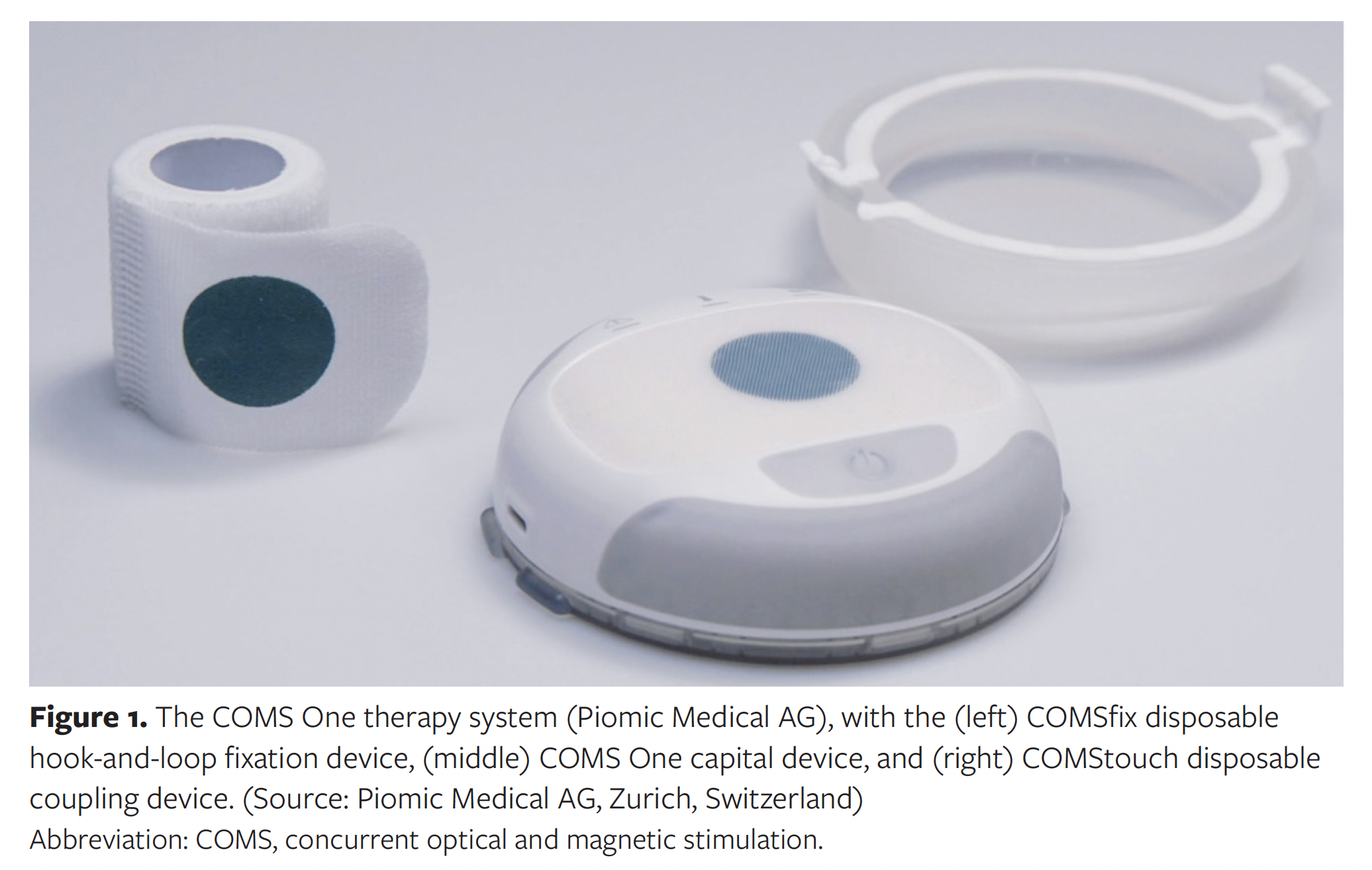
To administer a procedure with the COMS system, the capital device is first connected to the sterile coupler through a press-fit mechanism to form an integrated treatment assembly. This assembly is then placed against, and directly over, the patient’s ulcer. The sterile coupler comes in direct contact with the periwound skin, while the capital device, being mounted atop the coupler, does not. Once in position, the treatment assembly is secured to the patient using the fixation strap, which does not contact the wound. Patients are sedentary during the procedure. Therapy is started using a push button on the capital device. Once therapy is complete, the sterile coupler and fixation straps are removed and disposed. Figure 2 shows the positioning of the COMS system on the lateral mid- and forefoot of a patient who is not enrolled in the MAVERICKS trial.
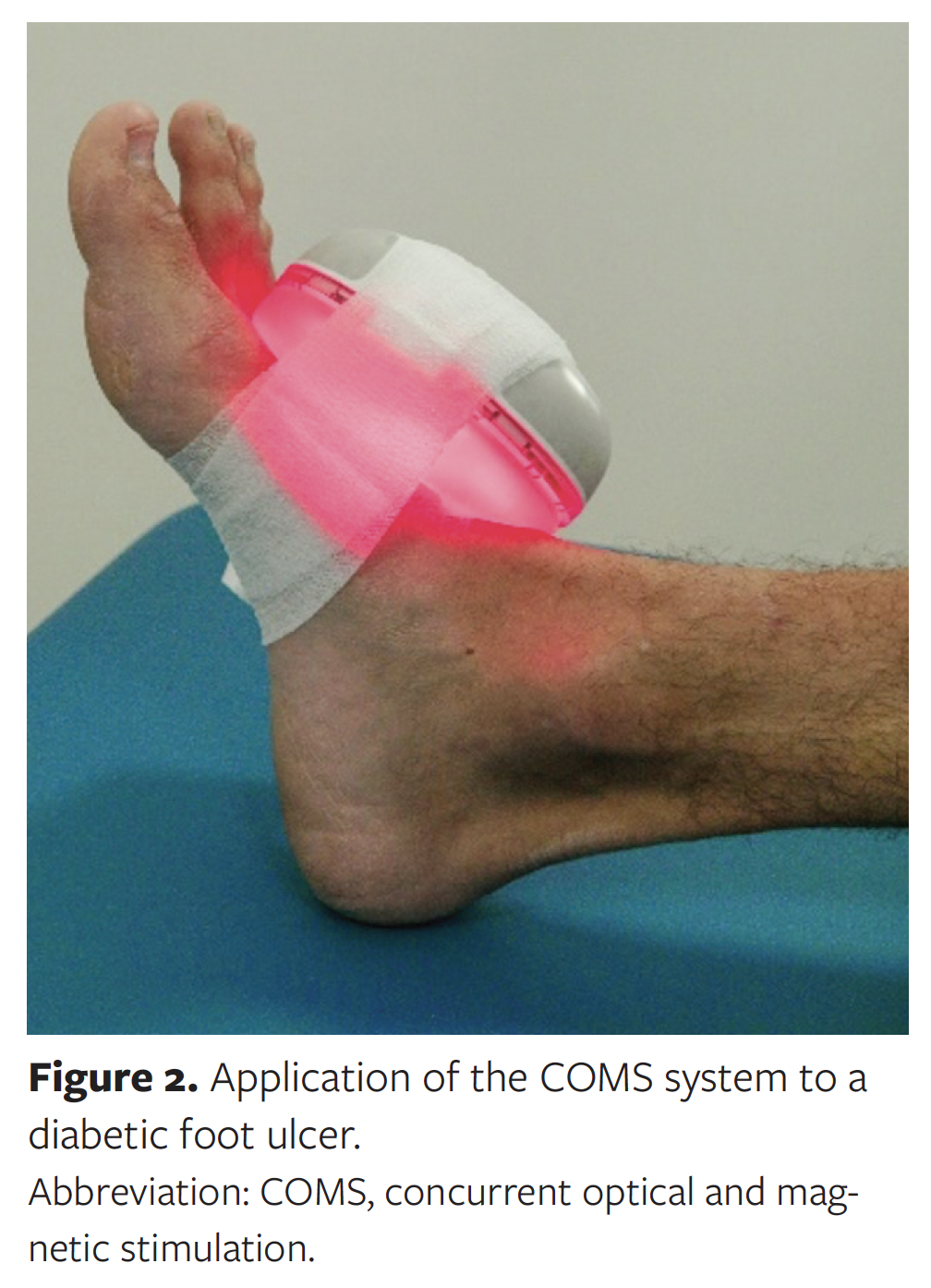
In the MAVERICKS trial, COMS treatments are delivered directly to the wound bed and periwound skin. Treatments are administered after removing existing dressings and preparing the wound bed. Debridement is performed if necessary to remove devitalized tissue, such as eschar and slough. There are no known topical agents that prevent the optical and magnetic stimulation generated by the COMS system from sufficiently penetrating the wound bed and periwound skin. Additionally, the COMS system operates in a nonreactive, low-energy light spectrum (>650 nm) and is not expected to interfere with or cause photosensitivity or photodegradation when used in conjunction with commonly prescribed antibiotics.
Qualified health care professionals eligible to administer COMS procedures during the trial include medical doctors, podiatrists (doctors of podiatric medicine), osteopathic physicians (doctors of osteopathic medicine), physical therapists, nurse practitioners, and registered nurses. However, after the trial’s completion, pending FDA marketing authorization and subsequent commercialization, COMS procedures can be administered under general physician supervision, allowing a significantly wider range of clinicians to deliver the therapy.
The COMS system received European Conformity (CE) certification in 2020 for DFU treatment in the European Union (EU) and has demonstrated promising preliminary clinical outcomes, including significant wound size reductions and improved healing rates.7 Preliminary studies have demonstrated consistently high efficacy outcomes, with wound size reductions averaging 69% and closure rates of 62% to 100% for specific ulcer types.8 These encouraging data justify further investigation of COMS as a potential breakthrough in DFU treatment.
Materials and Methods
Study design and purpose
The MAVERICKS trial is a multicenter, randomized, sham-controlled, double-blind pivotal study (FDA Category B IDE) designed to evaluate the COMS system as an adjunct to standard of care (SOC) for refractory DFUs. The purpose of this clinical trial is to evaluate the safety and effectiveness of treatment with the COMS system in subjects with refractory DFUs. The prospective, randomized, double-blind, sham-controlled trial is designed to demonstrate superiority of wound closure of the COMS system compared with a sham-control device through 24 weeks post-application, when each is administered in conjunction with SOC in the treatment of refractory DFUs. This primary end point aligns with the consensus reached at the 2022 FDA Public Workshop on Wound Healing Clinical Trial Design, where wound healing thought leaders and regulatory experts proposed for consideration that the traditional 12-week complete closure end point by itself may not adequately reflect real-world clinical outcomes.10 As a result, the FDA endorsed time to healing over 24 weeks as a clinically meaningful end point and supported its inclusion in the design of the current study.
The FDA categorizes IDE-approved devices based on whether available data demonstrate that initial questions of safety and effectiveness have been resolved. FDA IDE-approved devices are assigned to either Category A, experimental (initial safety and effectiveness questions not resolved), or Category B, nonexperimental or investigational (initial safety and effectiveness questions resolved).11 In this case, the FDA concluded that the COMS system resolved the initial questions of safety and effectiveness for this type of device, resulting in the MAVERICKS trial being approved as a Category B IDE (IDE number G220277).12 The pivotal EU trial that formed the basis for CE mark certification of the COMS system demonstrated that, in terms of device safety, no relevant issues were identified for clinical implementation, with adverse events (AEs) being mostly mild and transient, and resolving spontaneously.7 Most AEs occurred before COMS treatment began, and no autocorrelation was found, indicating that the COMS system did not increase AE incidence.
The trial involves up to 30 US investigational sites, representing a diverse range of clinical settings, including hospital outpatient wound centers, specialty wound clinics, physician practices, academic institutions, and Veterans Administration Medical Centers. All patient visits and treatments occur at the study site, where medical personnel administer all treatments. Principal investigators (PIs) from specialties such as podiatric surgery, vascular surgery, trauma surgery, plastic surgery, and family medicine oversee the study. The Centers for Medicare & Medicaid Services (CMS) approved the COMS system, as well as related and routine items and services, for Medicare coverage during the MAVERICKS trial.12 Additionally, CMS had direct input into the final MAVERICKS protocol through iterative discussions with the proprietary device manufacturer.
Trial population
A total of 450 subjects with refractory DFU will be screened. It is expected that 50% of subjects will be excluded from the trial if either of the following occurs between screening and randomization: greater than 30% wound closure during any 2-week interval, or greater than 50% wound closure over the total 4-week period (measured post-debridement). The remaining 224 subjects will be randomized into 2 groups, with 112 subjects undergoing treatment with the sham device and 112 subjects undergoing treatment with the active COMS device. The enrollment target of 224 patients is an intention-to-treat (ITT) population, and 10% dropout (lost to follow-up) of randomized subjects is expected. This clinical trial aims to provide significant evidentiary support for the COMS system, and the results will facilitate FDA marketing authorization through the De Novo regulatory pathway for novel medical devices. With greater than 50% of the cohort aged 65 years or older, the diverse patient base reflects real-world wound care delivery scenarios, enhancing generalizability to the broader Medicare population and further supporting the trial’s applicability for reimbursement. Upon either the aged younger than 65 years or the aged 65 years or older arm reaching the 50% enrollment threshold across all study sites, that arm will be closed to additional enrollment.
Trial duration
Subjects who are consented for this clinical trial will participate for a maximum number of 23 visits over 28 weeks, which includes 2 weeks to 4 weeks for screening, 12 weeks for treatment and observation, and 12 weeks for follow-up. The estimated enrollment period is 18 months. However, the trial will continue until all subjects planned by this protocol have been randomized. For this reason, the overall duration of the trial, from enrollment of the first subject to the last follow-up visit of the final subject, is estimated at 36 months.
COMS exposure duration
Subjects are exposed to COMS for a maximum of 12 weeks. During the 8-week treatment phase, the assigned device treatment (active or sham) is administered no more than twice a week. A minimum of 12 treatment applications must be delivered, unless wound closure occurs prior to the completion of all treatments. In the event of complete wound closure prior to conclusion of the observation phase, treatments will cease, and visits will decrease to once weekly. Wounds that remain open at the end of the observation phase are assessed for complete reepithelialization during the follow-up phase, during which subjects are seen once monthly until wound closure or their final trial visit. Figure 3 shows the study timeline and visit schedule.
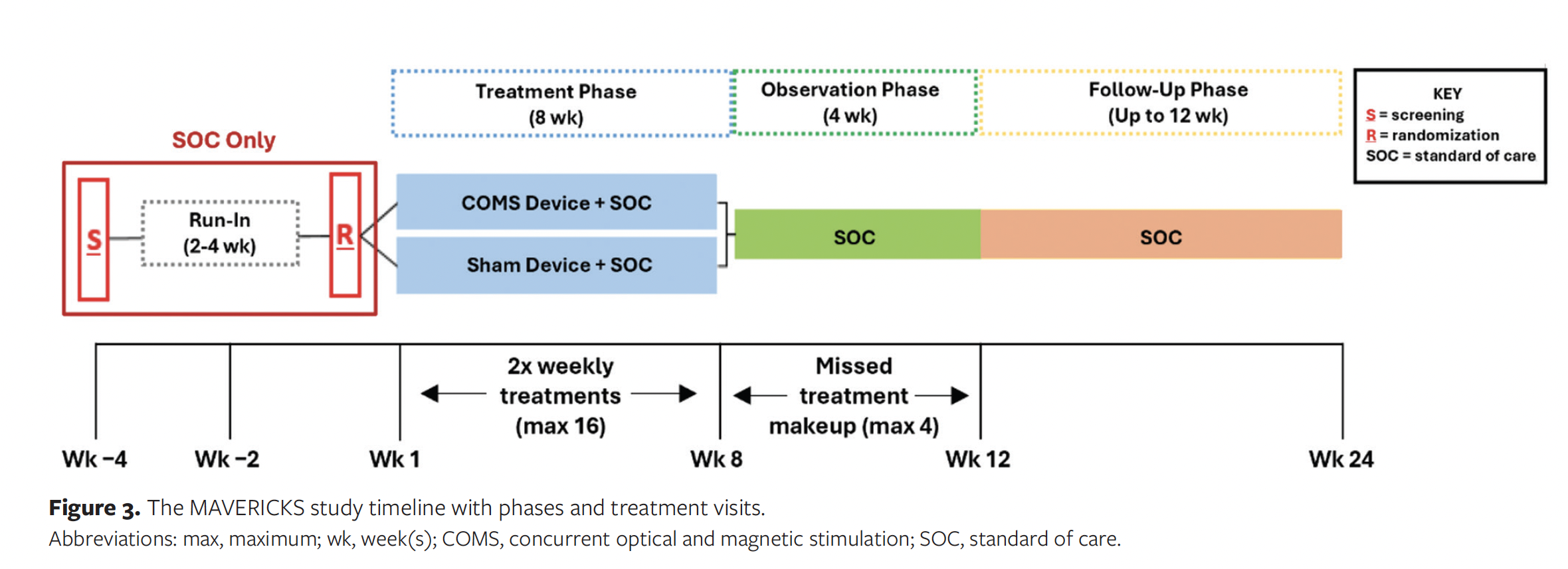
Outcomes
The primary outcome is the time to complete wound closure, defined as full skin reepithelialization without drainage, confirmed at 2 consecutive trial visits 2 weeks apart, within 24 weeks of randomization.
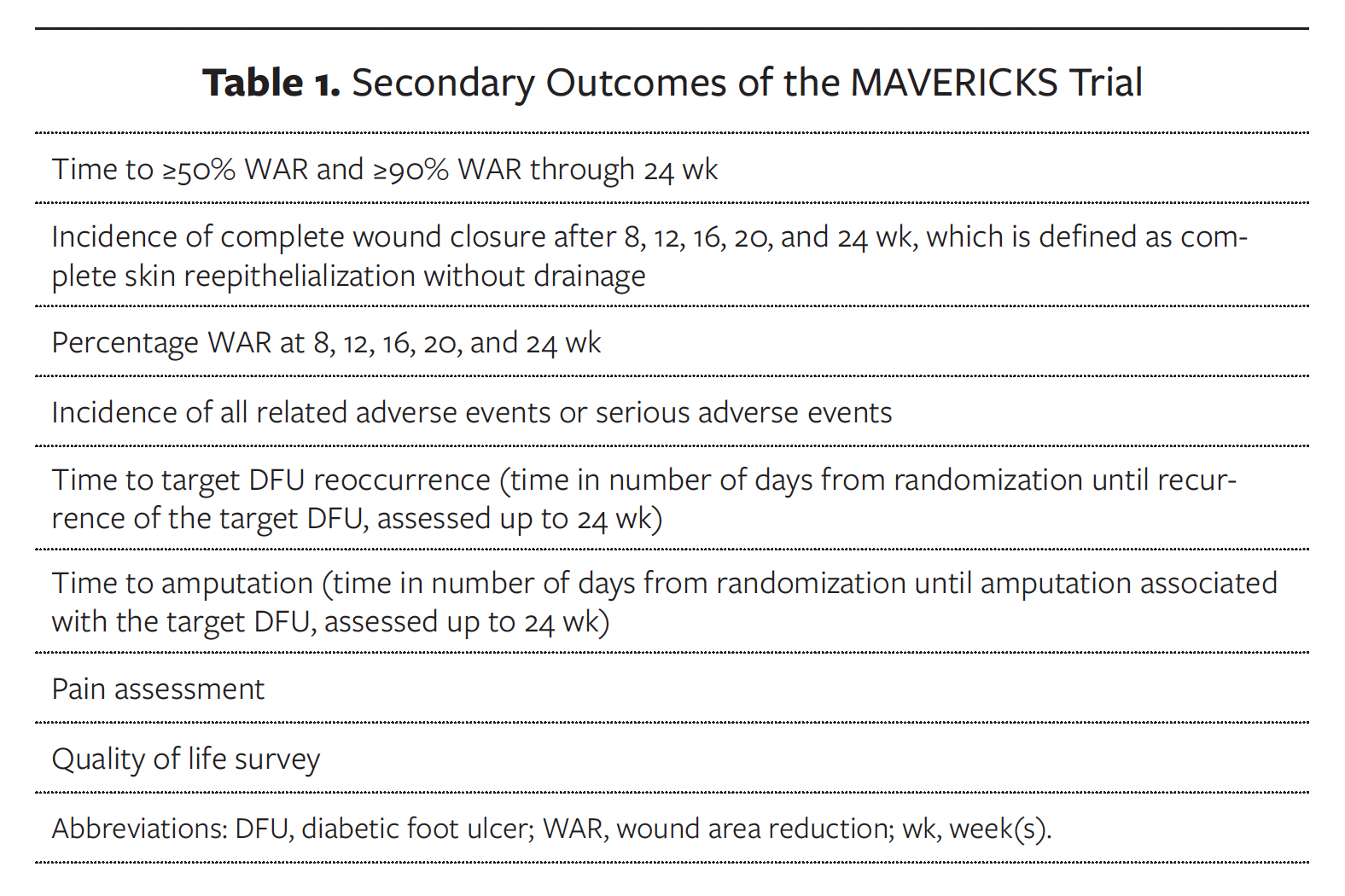
Secondary outcomes consist of assessment of wound healing parameters, determination of the proportion of subjects in both treatment groups with partial or complete wound closure at various time points, and confirmation of safety by analyzing AEs associated with COMS, the COMS system, and/or other trial procedures related to the management of DFUs. These outcomes are listed in Table 1.
Inclusion and exclusion criteria
Key inclusion criteria are patients aged 22 years to 90 years with type 1 or type 2 diabetes, a Wagner grade 1 or 2 DFU located at or below the malleoli, and ulcer duration of 30 days to 52 weeks. Eligible ulcers must measure between 0.5 cm² and 25 cm² post-debridement, with adequate vascular perfusion of the affected limb demonstrated by specific parameters such as skin perfusion pressure greater than or equal to 30 mm Hg, ankle-brachial index greater than 0.7 but less than 1.2, toe-brachial index greater than 0.4 but less than 1.1, or transcutaneous oxygen pressure greater than 40 mm Hg.
Exclusion criteria include pregnancy, active or historical skin cancer in treatment areas, systemic infections, osteomyelitis, dialysis dependency, recent systemic corticosteroid therapy exceeding specified thresholds, or significant laboratory abnormalities (eg, hemoglobin A1c >12%, serum creatinine >4.0 mg/dL). An osteomyelitis diagnosis is made by the treating physician through physical examination, generally accepted laboratory and clinical findings, imaging tools, histopathological and microbiological assessment of bone, or any combination thereof, as chosen by the PI or treating physician. Patients with a contralateral major amputation are not excluded from enrolling in the study, nor are patients with an ipsilateral minor amputation, provided that any target ulcer on the site of the minor amputation is determined to be a DFU and not an acute wound resulting from the prior amputation.
Screen failures
Subjects who do not meet the criteria for participation in this trial (screen failure) due to not meeting the inclusion and exclusion criteria may be rescreened 1 time. Subjects are required to wait a minimum of 30 days before being rescreened. Subjects who have a screen failure due to substantial healing or wound closure during the run-in phase are not eligible for rescreening with the same target DFU. Rescreened subjects will be assigned a different subject number from the one designated at their initial screening.
Run-in phase
To ensure a homogeneous population of refractory ulcers, a dual-track run-in phase is implemented to identify truly refractory ulcers. Participants with documented wound history undergo a 2-week assessment, while those without prior records complete a 4-week phase. Exclusion criteria during this phase include greater than 30% wound area reduction during any assessment interval or greater than 50% reduction across the run-in period. This stringent screening minimizes variability and ensures inclusion of ulcers that are unlikely to heal with SOC alone, which carry higher risks of nonhealing and complications (eg, amputation).
The extended run-in phase differentiates this trial from typical DFU studies, which often use shorter screening periods, resulting in greater variability and inclusion of patients who exhibit significant healing solely due to improved compliance with SOC.
Randomization and treatment protocol
Participants meeting all eligibility criteria are randomized using a random permuted block design stratified by investigational site and age group (<65 years or ≥65 years) with a 1:1 allocation ratio in either the sham control or active treatment group via iMedNet (Mednet). The control group receives a sham device that looks identical to the active device, including emitting red light on the perimeter but not to the wound. Neither the active nor the sham device creates palpable sensations through the delivery of electromagnetic stimulation that would affect blinding. The sponsor (Piomic Medical, Inc.) provided comparative images and detailed operational descriptions of the investigational and sham devices to the FDA as part of its IDE approval submission. The FDA determined that the devices are sufficiently indistinguishable in both appearance and behavior during operation, and consequently approved the blinding methodology. Prior to the first patient enrollment at any site, the medical device contract research organization, NAMSA, on behalf of the sponsor, will conduct a site initiation visit to provide, among other extensive training, hands-on COMS active and sham device training.
Replicating real-world treatment conditions, SOC during and between treatment visits is at the discretion of the treating physician, including the frequency and method of debridement, as well as the choice of wound dressings, antibiotics, and other routine medical supplies. In accordance with best practices, the study sponsor recommends that all patients clinically indicated for an off-loading boot receive one, and therefore the sponsor covers the cost of the device in the protocol. However, the specific off-loading boot is selected based on each individual patient’s needs.
Concomitant therapies
For this protocol, only prescription medications will be reported. Dietary supplements and over-the-counter medications will not be reported unless ordered specifically to treat the refractory DFU. Medication reporting will include start and stop dates, daily dosage of medication, and frequency. The concomitant therapies in Table 2 are not allowed during trial participation.

Study visits and assessments
Each visit follows a standardized protocol. At each study site, an assessor blind to treatment allocation evaluates the wound before any debridement or treatment, ensuring unbiased outcome assessments. The blinded assessor is a clinician designated by the PI who is qualified to perform wound assessments in compliance with the study protocol. The PI or a qualified clinician then performs wound debridement, followed by imaging and measurement using a proprietary digital wound imaging system (Swift Skin & Wound; Swift Medical, Inc). Data collected during visits include AEs, concomitant medications, and application of standardized wound dressings. Quality of life metrics are collected using the 36-Item Short Form Health Survey questionnaire, and pain assessments are recorded at specific intervals (weeks 1, 8, 12, and 24).
If wound closure is achieved before all 16 treatments are administered, participants continue scheduled visits for an additional 2 weeks to confirm sustained closure. Final verification occurs 2 weeks after the blinded assessor’s initial closure determination.
Photography and wound measurement
The proprietary digital imaging system is used to ensure accurate and consistent wound documentation. Adhesive markers calibrate the software for precise wound boundary measurements. A blinded physician employed by the manufacturer of the proprietary digital imaging system reviews every wound image to confirm accurate wound tracing and measurements and, if necessary, makes corrections. This dual-validation approach strengthens the reliability of both inclusion and exclusion decisions and outcome assessments.
Statistical methods and powering
The time to complete healing from randomization is the primary end point compared between treatment and control groups using a log-rank test for 2 independent survival curves. For subjects who do not achieve complete wound closure within 24 weeks of randomization, time to event will be censored at the last target DFU assessment or at randomization in the event of no target DFU assessments. The primary analysis set for all end point analyses and results will be the ITT analysis set, with all other analysis sets defined as supportive. Sensitivity analysis for all analyses will be performed using a modified ITT analysis set (patients who received at least 1 treatment) and a per-protocol analysis set. The study plans to enroll 224 randomized subjects. This is expected to provide a sample size of 202 randomized subjects with primary end point outcome data, after accounting for approximately 10% missing data due to early trial withdrawal or missed end point assessment. Accordingly, an approximate 85% power is anticipated for the primary effectiveness end point across the 2 planned analyses (1 interim and 1 final).
Discussion
DFUs, particularly those that prove refractory to SOC regimens, represent a significant clinical, economic, and social burden. DFUs affect up to one-third of individuals with diabetes and often lead to severe complications such as amputation and reduced life expectancy.13 Unfortunately, the 5-year mortality rate following major amputation is 82.6% greater than all reported cancers combined, highlighting the critical need for timely and sustained wound closure.14 The financial impact of DFUs is also substantial, with an estimated annual cost of $413 billion in 2022 for diabetes and its complications.15
Many advanced wound care therapies present a major challenge in DFU healing due to their high procedural complexity and associated cost. Cellular and/or tissue-based products (CTPs), platelet-rich plasma (PRP), and hyperbaric oxygen therapy are examples of commonly used but costly procedures, with CTPs and PRP being invasive as well.16,17 These treatments are usually administered in resource-intensive primary health care facilities by physicians or other qualified wound care providers. This can limit access for many patients, particularly those who are socially and economically disadvantaged.18 Efficacy data for some of these products are encouraging.19-21 However, the use of various advanced wound care therapies requires highly skilled clinical personnel and sufficient payer coverage. By comparison, COMS’s intuitive design and 16-minute treatment protocol require minimal instruction for effective utilization.8 User satisfaction scores reflect this ease of implementation and ongoing utilization, with ratings of 9.3 out of 10 for ease of use and 8.7 out of 10 for application confidence. Ease of use is based on a 10-point scale in which 0 equals “very hard to use” and 10 equals “very easy to use, while application confidence is also based on a 10-point scale in which 0 equals “very insecure application” and 10 equals “very confident application.”8
The MAVERICKS trial represents an advancement in the clinical investigation of hard-to-heal DFUs. Unlike many wound healing studies, it incorporates an extended 4-week run-in phase to identify and enroll only truly refractory ulcers, that is, those deemed unlikely to heal with SOC alone. This stringent preselection process ensures study focus on a homogeneous and challenging patient population, thereby addressing a critical gap in prior research. The authors of the current study are aware of only 1 other study that incorporated a similar run-in phase; in that trial by Game et al,22 only the independent wound healing assessor was masked to subject allocation. The participants and clinicians administering therapy were not blinded to treatment allocation, and no sham control was used, potentially introducing study bias. The MAVERICKS trial by comparison is an FDA-approved IDE study that strives for data integrity by incorporating a robust double-blind, sham-controlled methodology. The use of a sham device that is indistinguishable from the active treatment minimizes bias and ensures the highest level of scientific rigor. By combining these elements, the MAVERICKS trial sets a new benchmark for studies in advanced wound care, with an overriding goal to generate data of increased reliability. The full data set from the MAVERICKS study is expected by the end of 2027.
Limitations
Despite the regulatory oversight inherent in this FDA Category B IDE clinical trial, several limitations could affect generalizability. Enrollment is limited to patients with Wagner grade 1 or 2 DFU. While consistent with the device’s intended use, this may not fully represent real-world outcomes in patients with more complex ulcers. The trial includes more than 25 geographically diverse study sites in the United States, primarily hospital outpatient wound care departments and physician-led clinics. These are common settings for refractory ulcer treatment, but the absence of post-acute, long-term care, and home health settings may limit extrapolation to patients managed in those environments. As a Medicare-approved trial, enrollment criteria required that at least 50% of participants be 65 years of age or older, potentially weighting the study population toward older adults. Despite these limitations, the study’s well-structured and comprehensive design includes multiple controls that reinforce data integrity and reliability, setting it apart from much of the existing wound care research.
Conclusion
By using a sham-controlled, double-blind protocol design, the MAVERICKS clinical trial seeks to provide high-quality evidence on the safety and efficacy of COMS as an adjunctive therapy for hard-to-heal DFUs. The trial’s extended screening phase, comprehensive inclusion and exclusion criteria, and standardized treatment assessments position it to provide relevant data for the Medicare and general patient populations, while offering valuable insights to guide clinical practice.
By addressing critical clinical challenges with an innovative and accessible approach, the COMS system has the potential to transform DFU care. Its ability to integrate seamlessly across diverse health care settings while maintaining cost-effectiveness and reproducible clinical results may establish the COMS system as a vital tool to advance chronic wound management and improve patient outcomes. Upon trial completion, the research team looks forward to sharing the findings and outcome analysis with wound care stakeholders.
Trial status
This is an ongoing, multicenter pivotal clinical trial actively screening and enrolling subjects across multiple study sites in the United States. Patient recruitment began in June 2023 and is expected to be completed in 2026. Physicians or clinical centers interested in referring patients are encouraged to visit ClinicalTrials.gov (Identifier: NCT05758545; https://clinicaltrials.gov/study/NCT05758545), for additional trial information, including eligibility criteria and participating sites, as well as contact information for participating study sites and sponsor representatives.
Role of the funder and sponsor
Funding for the MAVERICKS study is provided by Piomic Medical AG, headquartered in Zurich, Switzerland, on behalf of the study’s sponsor, Piomic Medical, Inc, located in Rosemont, IL. The funder directed the design of the study and, in close collaboration with the sponsor and the study’s contract research organization, NAMSA, oversees data collection, site engagement, and overall trial management. Neither the funder nor the sponsor will be involved in data analysis or data interpretation. Preparation of the protocol and the decision to publish rest solely with the academic authors. While the funder and sponsor will have access to the final dataset, they lack authority to influence or prevent publication of the study’s outcomes. Patients and members of the public did not participate in the design of this study and are not involved in its ongoing conduct, data reporting, or dissemination planning.
Institutional Review Board statement
The study is approved by WCG Institutional Review Board (WCG IRB), 5000 Centregreen Way, Suite 200, Cary, NC, 27513, USA, 609-945-0101, Protocol number 20230014.
Safety oversight
An independent data safety monitoring board (DSMB) is composed of physicians who have expertise in the area of treating refractory DFUs and also includes a biostatistician. The members of the DSMB do not have any conflicts of interest and are not investigators in the clinical trial. The primary responsibilities of the DSMB include the following: (1) periodic review and evaluation of the accumulated trial data for subject safety, trial conduct and progress, and, when appropriate, validity and integrity, and (2) making recommendations to the study’s sponsor concerning the continuation, modification, or termination of the clinical trial.
An independent Clinical Events Committee (CEC) comprises physicians who have expertise in the area of wound management and diabetes ulcer care. The members of the CEC do not have any conflicts of interest and are not investigators in the clinical trial. The CEC independently reviews and adjudicates safety and/or effectiveness end points in a blinded, unbiased, confidential, and consensus-based manner. At a minimum, the CEC determines whether events meet protocol-defined end points and adjudicates events as either related to the trial device or concomitant therapy.
Ethics approval and consent to participate
The MAVERICKS clinical trial conforms to the US Food and Drug Administration regulations for the protection of human subjects (21 CFR Parts 50 and 56), investigational new drug application requirements (21 CFR Part 312), and the Health Insurance Portability and Accountability Act for the protection of participant privacy. The study also complies with the ethical principles described in the Declaration of Helsinki and the International Council for Harmonisation Good Clinical Practice guideline. WCG IRB reviewed and approved the protocol prior to study initiation. All participants (or their legal guardians, as applicable) provide signed informed consent before enrollment.
Author and Public Information
Authors: Robert D. Galiano, MD1; Rena A. Li, BA1; John C. Lantis, MD2; Alisha Oropallo, MD3; Jesus Ulloa, MD4; Mark Iafrati, MD5; Lawrence A. Lavery, DPM, MPH6; Jessica O’Connell, MD4; and Aksone Nouvong, DPM4
Affiliations: 1Northwestern University Feinberg School of Medicine, Department of Plastic Surgery, Chicago, IL, USA; 2Icahn School of Medicine at Mount Sinai, Department of Surgery, New York City, NY, USA; 3Donald and Barbara Zucker School of Medicine at Hofstra University/Northwell, Department of Vascular Surgery, Hempstead, NY, USA. Feinstein Institutes for Medical Research, Manhasset, NY, USA; 4David Geffen School of Medicine at University of California Los Angeles, Department of Surgery, Los Angeles, CA, USA; 5Vanderbilt University Medical Center, Department of Vascular Surgery, Nashville, TN, USA; 6University of Texas Health Science Center, Department of Orthopedic Surgery, San Antonio, TX, USA
Disclosure: The authors disclose no financial or other conflicts of interest.
Ethical Statement: The MAVERICKS clinical trial conforms to US FDA regulations and HIPAA for the protection of participant privacy. The study also complies with the Declaration of Helsinki. The WCG Institutional Review Board reviewed and approved the protocol, and all participants (or their guardians) provided informed consent.
Correspondence: Aksone Nouvong, DPM; David Geffen School of Medicine at University of California Los Angeles, Department of Surgery, 200 Medical Plaza, Suite 504, Los Angeles, CA 90095; anouvong@mednet.ucla.edu
Manuscript Accepted: July 17, 2025.
Recommended Citations
Galiano RD, Li RA, Lantis JC, et al. The trial design of the Concurrent Optical and Magnetic Stimulation (COMS) Therapy Study for refractory diabetic foot ulcers (MAVERICKS): a multicenter, randomized, sham-controlled, double-blind investigational device exemption clinical study. Wounds. 2025;37(8):275-282. doi:10.25270/wnds/25037
References
1. Holzer-Geissler JCJ, Schwingenschuh S, Zacharias M, et al. The impact of prolonged inflammation on wound healing. Biomedicines. 2022;10(4):856. doi:10.3390/biomedicines10040856
2. Schultz GS, Chin GA, Moldawer L, Diegelmann RF. Principles of wound healing. In: Fitridge R, Thompson M, eds. Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists. Adelaide (AU): University of Adelaide Press; 2011:103–108, 331–334.
3. Mo Y, Sarojini H, Wan R, et al. Intracellular ATP delivery causes rapid tissue regeneration via upregulation of cytokines, chemokines, and stem cells. Front Pharmacol. 2020;10:1502. doi:10.3389/fphar.2019.01502
4. Chiang B, Essick E, Ehringer W, et al. Enhancing skin wound healing by direct delivery of intracellular adenosine triphosphate [published correction appears in Am J Surg. 2008;195(1):139]. Am J Surg. 2007;193(2):213-218. doi:10.1016/j.amjsurg.2006.08.069
5. Hamid S, Hayek R. Role of electrical stimulation for rehabilitation and regeneration after spinal cord injury: an overview. Eur Spine J. 2008;17(9):1256-1269. doi:10.1007/s00586-008-0729-3
6. Koel G, Houghton PE. Electrostimulation: current status, strength of evidence guidelines, and meta-analysis. Adv Wound Care (New Rochelle). 2014;3(2):118-126. doi:10.1089/wound.2013.0448
7. Reinboldt-Jockenhöfer F, Traber J, Liesch G, Bittner C, Benecke U, Dissemond J. Concurrent optical and magnetic stimulation therapy in patients with lower extremity hard-to-heal wounds. J Wound Care. 2022;31(Suppl 6):S12-S21. doi:10.12968/jowc.2022.31.Sup6.S12
8. Neyens J, van Heusden W, Van Veenendaal D, Schols J. Effects of concurrent optical and magnetic stimulation in hard-to-heal wounds: a real-world evidence case series. J Wound Care. 2024;33(8):560-568. doi:10.12968/jowc.2024.0133
9. Traber J, Wild T, Marotz J, Berli MC, Franco-Obregón A. Concurrent optical- and magnetic-stimulation-induced changes on wound healing parameters, analyzed by hyperspectral imaging: an exploratory case series. Bioengineering. 2023;10(7):750. doi:10.3390/bioengineering10070750
10. Public Workshop: FDA Wound Healing Scientific Workshop, April 28–29, 2022. https://www.fda.gov/media/167141/download
11. FDA Categorization of Investigational Device Exemption (IDE) Devices to Assist the Centers for Medicare and Medicaid Services (CMS) with Coverage Decisions, December 5, 2017. https://www.fda.gov/media/98578/download
12. Centers for Medicare & Medicaid Services. Approved Investigational Device Exemption Studies. https://www.cms.gov/medicare/coverage/investigational-device-exemption-ide-studies/approved/g220277-nct05758545
13. Armstrong DG, Boulton AJM, Bus SA. Diabetic foot ulcers and their recurrence. N Engl J Med. 2017;376(24):2367-2375. doi:10.1056/NEJMra1615439
14. Armstrong DG, Swerdlow MA, Armstrong AA, Conte MS, Padula WV, Bus SA. Five year mortality and direct costs of care for people with diabetic foot complications are comparable to cancer. J Foot Ankle Res. 2020;13(1):16. doi:10.1186/s13047-020-00383-2
15. Parker ED, Lin J, Mahoney T, et al. Economic costs of diabetes in the U.S. in 2022. Diabetes Care. 2024;47(1):26-43. doi:10.2337/dci23-0085
16. Carter MJ, Fife CE. Counting the cost of cellular and/or tissue-based products in diabetic foot ulcers: is there a justifiable price limit per square centimeter?. Adv Wound Care (New Rochelle). 2025;14(4):181-187. doi:10.1089/wound.2024.0087
17. Magruder ML, Caughey S, Gordon AM, Capotosto B S S, Rodeo SA. Trends in utilization, demographics, and costs of platelet-rich plasma injections: a ten-year nationwide investigation. Phys Sportsmed. 2024;52(1):89-97. doi:10.1080/00913847.2023.2178816
18. Beeckman D, Cooper M, Greenstein E, et al. The role community-based healthcare providers play in managing hard-to-heal wounds. Int Wound J. 2023;21(1):e14402. doi:10.1111/iwj.14402
19. Dardari D, Piaggesi A, Potier L, et al. Intact fish skin graft to treat deep diabetic foot ulcers. NEJM Evid. 2024;3(12):EVIDoa2400171. doi:10.1056/EVIDoa2400171
20. Smith J, Rai V. Platelet-rich plasma in diabetic foot ulcer healing: contemplating the facts. Int J Mol Sci. 2024;25(23);12864. doi:10.3390/ijms252312864
21. Sharma R, Sharma SK, Mudgal SK, Jelly P, Thakur K. Efficacy of hyperbaric oxygen therapy for diabetic foot ulcer, a systematic review and meta-analysis of controlled clinical trials. Sci Rep. 2021;11;2189. doi:10.1038/s41598-021-81886-1
22. Game F, Jeffcoate W, Tarnow L, et al. LeucoPatch system for the management of hard-to-heal diabetic foot ulcers in the UK, Denmark, and Sweden: an observer-masked, randomised controlled trial. Lancet Diabetes Endocrinol. 2018;6(11):870-878. doi:10.1016/S2213-8587(18)30240-7



